






A Silent Epidemic of Nutritional Imbalance
Over seventy million Americans may have nonalcoholic fatty liver disease.1 The disease begins with the accumulation of fat within the cells of the liver, but can progress to inflammation, the development of scar tissue, and in some cases death from liver failure or cancer.2-4 Simple accumulation of fat within the liver generally proceeds without producing any overt symptoms, but it is not necessarily harmless. The liver regulates blood glucose and blood cholesterol levels, plays a critical role in burning fat for fuel, helps eliminate excess nitrogen, contributes to the metabolism of endocrine hormones, stores vitamin A, protects against infections, and detoxifies drugs and environmental toxins.
Any type of damage to the liver is thus likely to impact whole-body health. Indeed, fatty liver disease increases the risk of cardiovascular disease three-fold in men, fourteen-fold in women, and seven- to ten-fold in type-one diabetics.5-6 Fatty liver is thus a dangerous silent epidemic, and as we will see, it is likely caused by the overabundance of calorie-rich, nutrient-poor refined foods and the banishment of traditional sources of choline like liver and egg yolks from the modern American menu.
Relation to Obesity, Diabetes, and Insulin Resistance
Samuel Zelman, a medical doctor from Topeka, Kansas, published the first human case series connecting fatty liver disease to obesity in 1952.7 Zelman decided to examine a group of obese patients for fatty liver after observing the disease in a hospital aide who drank twenty or more bottles of Coca-Cola per day. This obscene amount of soda provided 1,600 calories of sugar and the caloric equivalent of a pint or more of whisky per day. Obese people were so hard to find at the time that it took Zelman a full year and a half to find twenty obese people who were not alcoholics. All of his subjects showed some degree of liver damage, and about half had a significant degree of fatty liver disease.
Zelman knew that fatty liver occurred in the earliest animal models of obesity involving genetics or surgical damage to the hypothalamus,8-9 and suggested that hypothalamic damage, biological inheritance, and “psychological factors” contributed to obesity in humans. He proposed that obesity increased the caloric requirement and led to cravings for energy-dense, nutrient-poor foods that increased the need for choline and B vitamins. These foods then overloaded the liver with energy but failed to provide the nutrients needed to process that energy, resulting in fatty liver.
Zelman’s obese subjects universally preferred a diet rich in carbohydrate and fat but poor in protein. The one exception was a man who “related a fantastic story of fondness for the ingestion of huge beefsteaks,” but Zelman distrusted his story because the man’s psychiatrist had accused him of being a pathological liar. Zelman prescribed a diet containing “as much protein as possible,” emphasizing beef liver, lean fish, and lowfat dairy products such as skim milk and cottage cheese, as well as extra B vitamins and choline.
Zelman was likely on the right track when he identified nutrient density as an important component of a therapeutic diet, but his opposition to both fat and carbohydrate likely contributed to his regimen’s ultimate lack of success. He reported “as much difficulty and discouragement in dealing with the craving for carbohydrate- and fat-rich foods manifested by the obese as with the craving for alcohol of the chronic alcoholic.” Only two of his twenty subjects lost weight. He retested them for fatty liver and found some improvement, but he never reported completely successful resolution of obesity in any of his patients and consequently failed to document the success of his program in treating fatty liver.
Although type-one diabetes is much less common than obesity, it had been connected to fatty liver disease as early as 178410 and the resolution of diabetic fatty liver was an early objective of insulin therapy.11 Physicians identified the connection between alcohol consumption and fatty liver disease in the 1830s,12 however, and in the middle of the twentieth century the connection to obesity and diabetes was lost from mainstream consciousness as alcohol came to be seen as the exclusive cause of the disease. At least as early as the 1970s, this exclusive association of fatty liver with alcoholism had become so ingrained that physicians simply accused their patients of lying if they presented with fatty liver but denied drinking alcohol.13
In 1980, Jurgen Ludwig and other physicians from the Mayo Clinic produced a report of twenty patients with nonalcoholic fatty liver disease.13 Ninety percent of the patients were obese, 35 percent had heart disease, 30 percent had gall bladder problems, and 25 percent had type-two diabetes. These physicians coined the term “nonalcoholic steatohepatitis,” to describe the disease and abbreviated it “NASH.” The creation of a specific term for the nonalcoholic manifestation of the disease promoted greater awareness of this form, facilitated the organization of an entire research field devoted to its study, prevented doctors from accusing patients of lying about their alcohol intake, and, in the words of the Mayo Clinic physicians themselves, spared doctors “the embarrassment (or worse) that may result from the ensuing verbal exchanges.”
Since the Ludwig group’s landmark paper, numerous studies have confirmed the relation between fatty liver, obesity and diabetes. Autopsy14-15 and ultrasound16-17 studies have shown that rates of fatty liver are five- to fifteen-fold greater in obese individuals than in lean individuals and that the disease is present in up to three quarters of obese people. Similar studies have shown that 45 percent of type-one diabetics and 70-85 percent of type-two diabetics have fatty liver.6, 18-19 Moreover, even in the absence of diabetes and obesity, those with the lowest insulin sensitivity have the highest accumulation of liver fat.20
| SIDEBAR: THE FUNCTIONS OF THE LIVER The liver is the largest gland in the human body, accounting for about 2.5 percent of total bodyweight; it plays a large number of roles in promoting the health of the entire body. The liver regulates blood glucose and cholesterol levels, contributes to fat-burning, helps eliminate excess nitrogen, filters pathogens and particulate matter that would otherwise enter the bloodstream, detoxifies drugs and environmental toxins, and promotes the elimination of toxic chemicals and metals from the body.The liver is divided into four lobes, which include the left and right lobes as well as two smaller lobes called the quadrate and caudate lobes. Just under the right lobe lies the gall bladder, which acts as a reservoir that holds up to two to three tablespoons of bile. The blood supply to the liver is unique in that it comes from two sources. Blood that is rich in nutrients but relatively poor in oxygen flows from the stomach and intestine to the liver through the portal vein. This constitutes 60 to 70 percent of the blood that reaches the liver. Oxygen-rich blood from the lungs flows to the liver through the hepatic artery, which constitutes the remainder. These vessels infuse the liver tissue with blood through a network of large, irregular capillaries called sinusoids, until the blood finally leaves the liver through the central vein.Almost 95 percent of the cells in the liver are hepatocytes, which carry out the basic metabolic functions of the liver. The lining of the sinusoid itself is composed of sinusoidal endothelial cells, which are separated by small gaps that allow the passage of nutrients from the sinusoid to the hepatocytes. Several other cell types abide within the sinusoids, including stellate cells, Kupffer cells and a small number of pit cells. Stellate cells store vitamin A and lay down materials of the extracellular matrix such as collagen. Kupffer cells and pit cells are both members of the immune system. Kupffer cells are macrophages that function to remove bacteria and their byproducts, viruses, and undesirable particulate matter from the blood. Pit cells are natural killer cells that form a defense against viruses and tumors.In the fasting state, or when eating a high-fat, low-carbohydrate diet, we derive much of our energy from fats. The liver is responsible for converting fats into ketones and sending them into the blood so that other tissues may use them for energy. The liver also processes amino acids into glucose and disposes of excess nitrogen left over from this process as ammonia. In the fed state, however, and especially when we have eaten carbohydrates, insulin causes our liver to stop making ketones and glucose and to synthesize triglycerides instead, package them into very low-density lipoproteins (VLDLs), and send them into the bloodstream. When humans become insulin resistant, however, these processes go awry. The liver continues to synthesize triglycerides in response to insulin, but stops suppressing the synthesis of glucose. As a result, blood glucose levels rise, and the pancreas must secrete extra insulin to keep them under control. This extra insulin causes the liver to make even more triglycerides. The end result is poor control of blood glucose and elevated blood triglycerides. The liver closely ties together the regulation of blood lipids with the production of bile. Although the liver produces less than a sixth of the body’s cholesterol, it is the primary organ that expresses the LDL receptor, which takes up cholesterol-rich LDL particles from the blood. This not only lowers the cholesterol concentration in the blood, but also protects delicate components of the LDL particle from oxidation by ensuring that the particle is taken into cells before its stores of antioxidants run out. The liver then breaks down LDL particles and uses the cholesterol within them to synthesize bile acids, which promote the absorption of fat-soluble nutrients in the intestine. Thyroid hormone accelerates this process by signaling to the liver that the body is in a state of abundance and thus directing it to increase its expression of the LDL receptor, while many natural plant substances accelerate this process by causing the excretion of bile acids and causing the liver to divert more cholesterol into bile acid production. Contrary to the popular misconception that the liver “stores toxins,” its actual function is to process toxins into forms that can be more easily excreted. It mixes these processed toxins into the bile, together with toxic metals, glutathione, and many other substances before sending the bile out into the intestine. Glutathione protects the intestines from oxidative damage and is efficiently reabsorbed. We excrete most toxins that are packaged into the bile through either the feces or urine, but there are some “poorly behaved toxins” that constitute exceptions. For example, methyl mercury and arsenic are efficiently reabsorbed and circulate through the body repeatedly. Diclofenac, a non-steroidal anti-inflammatory drug (NSAID), uses its excretion into the bile as an opportunity to cause intestinal damage. By and large, however, the excretion of toxic drugs, metals, and environmental chemicals into bile means their safe elimination from the body. The liver plays many other important roles, including the metabolism of steroid hormones and the partial activation of vitamin D to 25-hydroxycholecalciferol or 25(OH)D. It should be clear from these many roles that damage to the liver of any form could potentially contribute to the development of many other diseases. Thus, ensuring proper liver function is imperative to supporting whole-body health. REFERENCES: Luxon, BA. Anatomy and Physiology of the Liver and Biliary Tree. In: Bacon BR and Di Bisceglie AM, eds. Liver Disease: Diagnosis and Management. Philadelphia, PA: Churchill Livingstone (2000) pp. 3-15.; Jaeschke H. Toxic Responses of the Liver. In: Klaassen CD, ed. Cassarett and Doull’s Toxicology: The Basic Science of Poisons: 7th Edition. New York, NY: McGraw-Hill Proffessional (2008) pp. 557-582. |
Since nonalcoholic fatty liver disease was rarely diagnosed before 198013 and since even today most people with fatty liver are probably not diagnosed,21 there is no way to track its prevalence over time. The prevalence of diagnosed diabetes in the United States, however, has increased from five to eight percent since 1988,22 and the prevalence of obesity has increased from about 15 percent to 35 percent since 1960.23 Given the high prevalence of fatty liver in obese and diabetic populations, we have likely experienced the emergence of a silent epidemic of fatty liver disease as the prevalence of obesity and diabetes has grown over the last few decades to reach epidemic proportions.
Diagnosing Fatty Liver Disease
Biopsy, ultrasound and magnetic resonance spectroscopy (MRS) are all legitimate methods of diagnosing fatty liver,6, 16-19, 21, 24-25 although ultrasound can sometimes fail to detect moderate cases of fat accumulation. Even though physicians often refer patients to biopsy when they have elevated liver enzymes,24 nearly 80 percent of patients with fatty liver have normal levels of these enzymes.21 In fact, in ten women who recently developed liver problems on an experimental diet low in choline, nine developed fatty liver but only one developed elevated liver enzymes.26 Fatty liver should therefore be suspected on the basis of obesity, diabetes and insulin resistance rather than elevated liver enzymes.
Fatty Liver As a Nutritional Imbalance
A well-functioning liver supports our health in many different ways (see sidebar). It is therefore deeply concerning that fatty liver is an independent risk factor for cardiovascular disease,5-6 as this would suggest that the disease may in fact compromise liver function and contribute to other degenerative diseases. Learning how to prevent and treat fatty liver, then, is critical.
In order to understand how our livers get fat, we must first realize that fatty liver disease occurs in two distinct stages (see sidebar for more detail). In the first, called simple steatosis, fat accumulates within the cells of the liver. In the second stage, inflammation, the proliferation of fibrous connective tissue (fibrosis), and eventually the formation of scar tissue (cirrhosis) ensue. In modern terminology, “nonalcoholic fatty liver disease (NAFLD)” refers to the full range of these disease states, while “NASH” refers only to the inflammatory stage.27
A number of experimental diets are currently used to study nonalcoholic fatty liver disease in laboratory animals, including diets high in fat, high in fructose, or deficient in choline and methionine. None of these models fully resembles the human situation, probably because they all emphasize single factors carried to extremes, whereas the human situation reflects a combination of several contributing factors (see sidebar).
The totality of the evidence suggests that the initial accumulation of fat in the liver is triggered by nutritional imbalance. As Zelman suggested in the 1950s, fatty liver seems to occur as a result of too much energy flowing through the liver without sufficient nutrients to process it. The accumulation of delicate fats, especially polyunsaturated fatty acids (PUFAs), increases the vulnerability of the liver to oxidative and inflammatory insults in the form of infections, toxins, or poor metabolism. These insults launch the progression from the first stage of simple fat accumulation to the second stage of inflammation.
The key culprits, then, are nutrient-poor refined foods, choline deficiency and polyunsaturated oils. The interaction between these factors can be seen by turning to the history of the development of animal models for fatty liver disease.
The Role of Refined Foods
In 1924, George Burr joined the laboratory of Herbert Evans, where Evans and Katherine Scott Bishop had recently discovered vitamin E.66 Evans and Bishop were having trouble reproducing their vitamin E-deficient diet, and Burr helped them develop a highly purified diet based deficiency that vitamin E could not cure, which Burr and his wife Mildred later identified as essential fatty acid (EFA) deficiency.67-68 Observing the fact that the annual per capita consumption of sugar in the United States had tripled over the preceding decades from 38 pounds to 115 pounds, Clarence Martin Jackson conducted a comprehensive analysis of the anatomy and tissue characteristics of rats fed Burr’s EFA-sufficient, 80 percent sucrose control diet.69 He compared them to rats fed 45 percent sucrose or 45 percent starch. Neither the 45 percent sucrose diet nor the 45 percent starch diet produced fatty liver, but the 80 percent sucrose diet produced moderate to severe cases of the disease. He noted that the liberal provision of cod liver oil, dried yeast and wheat germ satisfied the nutritional needs of the rats in all treatment groups, and that smaller amounts of sucrose may contribute to fatty liver in humans consuming nutritionally deficient diets. Indeed, dietary protein, methionine, and choline were later shown to protect against sucrose-induced fatty liver.12
In 1977, the American Institute of Nutrition (AIN), the principal professional organization for nutritional research scientists in the United States, developed standards for cereal-based, purified and chemically defined rodent diets.70 The purpose of the purified and chemically defined diets was to standardize diets between studies, so that toxicologists could easily make comparisons between one study and another. The days of cod liver oil, yeast and wheat germ were over. The days of purified vitamin and mineral mixes were now ushered in.
The AIN initially designated the purified diet as “AIN-76,” but they increased the vitamin K concentration ten-fold three years later in response to reports of excessive bleeding.71 They designated the new diet “AIN-76A.” Both of these diets were 50 percent sucrose and 15 percent starch, much lower in sucrose content than the diet that Jackson had used to induce fatty liver just a few decades earlier. Nevertheless, reports quickly began to surface of fatty liver developing spontaneously in rodents fed the AIN-76A diet.72 Reducing the concentration of sucrose from 50 percent to 20 percent resolved the fatty liver. Because of this and several other adverse metabolic effects of sucrose, the AIN released a new diet in 1993 and reduced the sucrose content to 10 percent, with the remainder of the carbohydrate supplied by cornstarch and a small amount of dextrinized cornstarch to aid in pelleting.73-74 As a result of many other problems that had surfaced with the diets, the AIN also increased the amount of vitamins E, K and B12, increased the calcium-to-phosphorus ratio, substituted soy oil for corn oil, and added various trace minerals not yet known to be essential.
| SIDEBAR: The “Two-Hit ” Hypothesis of Nonalcoholic Fatty Liver Disease The fact that some people who develop simple accumulation of fat in their livers, called steatosis, never develop the inflammatory form of fatty liver, called NASH,28 led to the “two-hit hypothesis.”29 This hypothesis considers steatosis to represent the results of a “first hit” that may itself be relatively benign, but nevertheless increases the vulnerability of the liver to a “second hit” in the form of oxidative or inflammatory insults that trigger the progression to NASH.This concept is supported by studies showing, for example, that genetically obese laboratory animals develop steatosis, but only develop strong evidence of NASH if they are treated with an inflammatory bacterial byproduct called endotoxin.30 Vitamin E, a fat-soluble antioxidant, is able to block this effect,31 showing the close relationship between inflammation and oxidative stress.The “first hit” is likely caused by increased synthesis of fat from carbohydrate,32-33 increased release of free fatty acids from fat stores into the blood stream,34 decreased burning of fat for fuel,35 and impaired export of triglycerides from the liver.36 These all lead to the accumulation of fat in the liver. The “second hit” involves the oxidative destruction of this fat, called lipid peroxidation.31, 37 The mere accumulation of oxidizable fat within the liver itself may increase the risk of lipid peroxidation.38 Impairment of mitochondrial function,39-42 spillover of fatty acids from mitochondria to other sites of fat-burning that burn fat in a much “dirtier” way,43-44 and inflammation30 further contribute to the development of NASH. Although some studies have failed to document progression from steatosis to NASH,28 others have documented a high rate of progression.2 A study published last year tracked people with steatosis for three years.2 A little less than half either stayed the same or got better, but almost 40 percent developed borderline NASH and over 20 percent developed fullfledged NASH.Simple steatosis may not be as benign as it appears. Among type-one diabetics in one study, those with steatosis were seven- to ten-fold more likely to have cardiovascular disease (CVD), and steatosis correlated with CVD independently of traditional risk factors.6 Another study tracked apparently healthy Japanese for five years.5 Those with steatosis at the beginning of the study were almost four times more likely than those without steatosis to develop CVD by the end of the study. When the researchers looked at the effect in men and women separately, the risk was increased three-fold in men and fourteen-fold in women. Remarkably, when they combined fatty liver into a statistical analysis with “metabolic syndrome”—a hodge-podge of symptoms including abdominal obesity, high blood pressure, elevated fasting glucose, elevated triglycerides, and low HDL-cholesterol—only fatty liver significantly predicted the risk of CVD. These studies do not prove cause and effect, but they do suggest the possibility that fatty liver deranges whole-body metabolism and contributes to negative health effects far beyond the boundaries of the liver itself. |
Why did sucrose prove so much more harmful in the context of the purified AIN-76 diet than it did when Jackson provided it in combination with cod liver oil, yeast, and wheat germ? In all likelihood, the provision of these unrefined foods supplied a wide variety of interacting vitamins, minerals, and other nutritional substances that aided in the metabolism of the sugar, helping the liver to burn it for energy, store much of the excess as glycogen, and export any fat made from it into the bloodstream. We will never know the exact nutritional composition of Jackson’s diet. We do know from other studies, however, that supplying extra choline in the diet provides powerful protection against fatty liver, whether induced by sugar, alcohol, or fat.
The Role of Choline
The discovery that choline could prevent the accumulation of fat in the liver was a byproduct of the seminal animal research conducted during the 1920s and 1930s showing that type-one diabetes was a disease of insulin deficiency. Physiologists first identified the role of insulin deficiency in type-one diabetes by studying the disease in dogs. In 1889 they produced diabetes by simply taking out the whole pancreas from these dogs and, after scrambling for a couple decades to identify the active component, they cured the diabetes with insulin in the early 1920s.75
| SIDEBAR: Animal Models of Fatty Liver Disease Researchers use a number of experimental diets to induce fatty liver in animal models, but none of them perfectly replicates the disease as it occurs in humans.HIGH-FAT DIETS : Rats consuming about 45 percent fat from coconut oil or butter do not develop steatosis even after fourteen weeks.45 Diets composed of 60 percent fat made mostly from lard cause steatosis to develop within eight weeks, but produce little evidence of NASH.46 Diets containing 70 percent fat made mostly from corn oil administered by gavage,47 intragastrically,48-49 or in liquid form50 induce clear evidence of NASH. In fact, a corn oil-based liquid diet will induce NASH in as little as three weeks.50 It is difficult to directly compare these studies because of the obvious differences in study design, but they do seem to suggest that corn oil is particularly noxious. Indeed, in nutrient-deficient and alcoholic models of fatty liver, replacement of corn oil with beef fat, cocoa butter or medium-chain triglycerides proves protective.51-53 Substitution of palm oil for fish oil also proves protective in the alcoholic model.54 High-fat diet models of fatty liver disease resemble the human situation in that they induce obesity,46 insulin resistance,50 and the spillover of fatty acids from the mitochondria to other sites where the fatty acids burn much “dirtier.”50 Their emphasis on polyunsaturated oils, moreover, resembles the great increase in linoleic acid and slight decrease in saturated fat that has occurred in American diets as the epidemic of fatty liver has emerged55-56 and the higher intake of omega-6 fatty acids found in NASH patients.57 These diets, however, are so high in fat that they suppress the synthesis of fat from carbohydrate, which is actually elevated in humans with fatty liver disease.33HIGH-FRUCTOSE DIETS : Diets containing 60 percent fructose cause increased blood triglycerides, insulin resistance, and steatosis in mice within five weeks.58 Over the course of 16 weeks, diets containing 30 percent glucose and 30 percent fructose will even induce mild inflammation.59 These models resemble the human situation in that synthesis of fat from carbohydrate is increased, and resemble the 30 percent increase in total fructose that has occurred since the introduction of high-fructose corn syrup60 as well as the statistical association between sweetened soft drinks and fatty liver.61-62 These diets, however, fail to induce obesity. They are very ineffective at inducing NASH, and what little inflammation does occur primarily occurs where blood enters through the portal vein,59 while in humans inflammation tends to occur where blood exits through the central vein.13METHIONINE-AND CHOLINE-DEFICIENT (MCD) Diets : Severe deficiency of methionine and choline impairs the ability of the liver to export triglycerides into the blood stream, which also occurs in humans.36 The deficiencies are so severe, however, that they induce weight loss instead of obesity. Although they make the liver insulin resistant, they actually increase whole-body insulin sensitivity.63-64 Nevertheless, MCD diets are useful for demonstrating the combined effects of different models. MCD diets are not just deficient in methionine and choline. They are usually loaded with sugar and corn oil as well. Fructose, as a component of sucrose, is required for the development of steatosis,65 while corn oil is required for the progression to NASH.37 Unlike the model using fructose alone, the inflammation occurs in a pattern consistent with human fatty liver.37 The experimental model that would most closely resemble the human situation would be a combination of sugar, polyunsaturated fat, and a much more moderate form of choline deficiency. |
Although cured of diabetes, the insulin-treated dogs nevertheless developed severe fatty liver degeneration and ultimately died of liver failure. Adding raw pancreas to their diet, which was composed of lean meat and sucrose, cured the problem. As researchers attempted to discover what it was in raw pancreas that cured the disease, they found in the early 1930s that egg yolk lecithin, which is abundant in choline, could cure it.76 Then they found that choline alone could cure it.77
It later turned out that the dogs became deficient in choline and methionine without a pancreas because they were not producing the digestive enzymes needed to free up those nutrients from the foods they were eating. Thus, simply providing them with the digestive enzyme trypsin could cure the fatty liver.78
In 1932 a group of researchers decided to replicate the fatty liver seen in the dogs in a nondiabetic rat model. What better way to stuff their livers with fat? Feed them fat! It seemed simple enough and it did indeed work. Although they had trouble reproducing the fatty liver with different colonies of rats or during the summer heat, they produced fatty liver in certain colonies of rats during the winter by replacing 40 percent of their ordinary cereal-based diet with beef drippings. Choline-rich lecithin derived from egg yolk or beef liver or simply choline itself cured the disease.79-80
Another group of researchers, however, tried to replicate this experiment in a group of rats who were consuming sufficient protein to maximize growth.81 They thus fed them 40 percent beef drippings but replaced another 20 percent of their cereal grains with the milk protein casein. This experiment failed miserably (Figure 1a). The researchers suspected that the casein might have been the problem, and they were indeed correct: on a choline-free, 40 percent beef dripping diet, reducing the casein from 20 to 5 percent doubled the level of fat in the liver (Figure 1b).82
We now know that choline is necessary to produce a phospholipid called phosphatidylcholine. This is a critical component of the VLDL particle, which we need to make in order to export fats from our livers. The amino acid methionine can act as a precursor to choline. Thus, the combined deficiency of choline and methionine will severely impair our abilities to package up the fats in our livers and to send them out into the bloodstream.83 This explains why casein was so effective at preventing fatty liver: it provided the rats with methionine that they could use to make choline.
Similarly, in 1949 a group of researchers showed that sucrose and ethanol had equal potential to cause fatty liver, and that increases in dietary protein, extra methionine and extra choline could each completely protect against this effect.12 Over fifty years later, in our own decade, researchers have shown that choline deficiency likewise causes fatty liver in humans.84 Thus, choline eventually proved capable of preventing fatty liver regardless of whether it was induced by feeding sugar, fat, or alcohol. These studies suggested that virtually any form of energy delivered to the liver can cause the accumulation of fat, so long as key nutrients needed to metabolize that energy—such as choline—were missing.
The Role of Polyunsaturated Oils
The initial experiments showing the protective effect of casein suggested that long-chain saturated fats might be more problematic than unsaturated fats or medium-chain fats (Figure 2a). As it turns out, saturated fats actually increase the need for choline slightly more than polyunsaturated fats (Figure 2b).85 Why this happens is unclear, but it may be that the body is quick to burn polyunsaturated fats for energy, since having them hang around is so dangerous. After all, if they hang around, they are likely to contribute to oxidative damage.
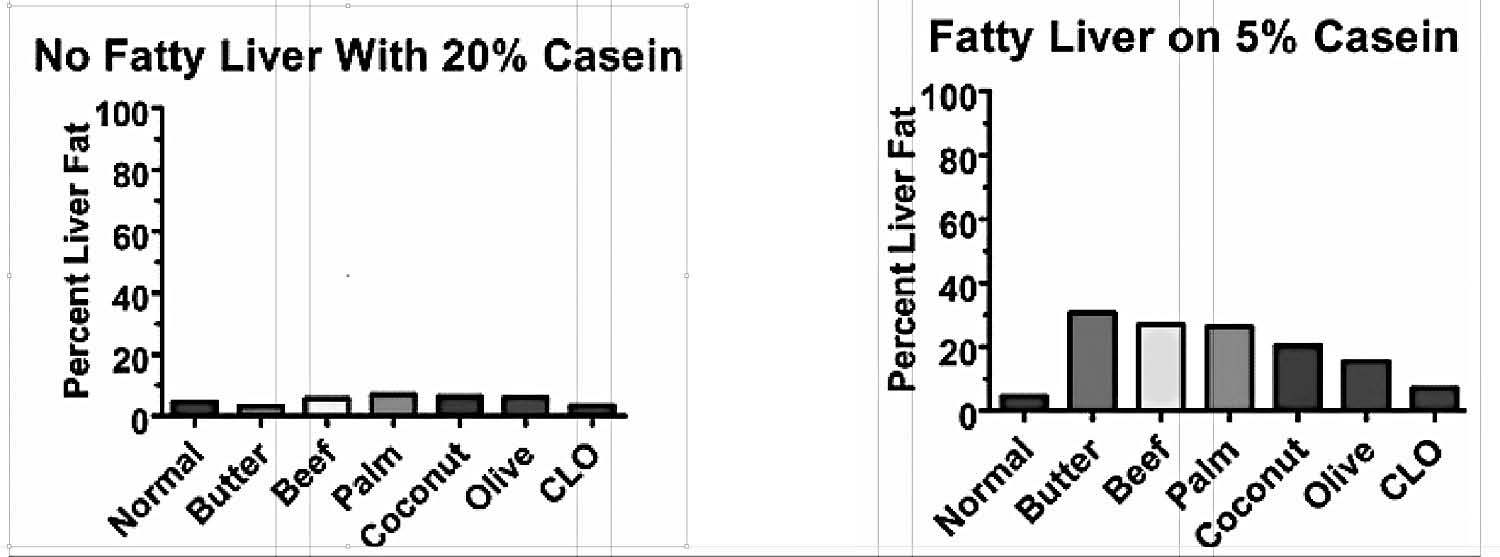
Figure 1. A. When rats were fed 20 percent casein, none of the different fats tested brought the level of fat in the livers above normal. B. When the amount of casein was reduced, fatty liver ensued. From references 81 and 82.
There is some experimental evidence suggesting that polyunsaturated fats may impair the export of fats from the liver by facilitating oxidative damage of the proteins involved.86 This evidence comes from isolated cells and live animals injected with fatty acids. Substituting coconut oil for corn oil prevents steatosis in some,52 but not all,37 studies using choline-deficient diets. Polyunsaturated oils are probably most likely to contribute to the accumulation of liver fat when combined with other factors that favor oxidative stress such as alcohol abuse, iron overload, toxin exposure, and poor metabolism.
There is much stronger evidence that polyunsaturated oils are responsible for the progression from simple steatosis to NASH.37 Substitution of carbohydrate, coconut oil or beef tallow for corn oil all prevent the oxidative damage and inflammation that results from methionine and choline deficiency during this stage of the disease.37 High-fat diets, moreover, only cause overt NASH when they are based on corn oil (see sidebar). These effects may result from a high intake of total PUFA, or may result from a high ratio of omega-6 to omega-3 fatty acids. Both of these factors are likely to play a role.
A Combination of Factors
As nonalcoholic fatty liver disease has emerged over the last several decades, refined foods have become commonplace. Liver has virtually disappeared from the American menu, and eggs have fallen victim to the anti-cholesterol campaign. These foods are the principal sources of choline (see Table 1). Total fructose intake has increased 30 percent, and intakes of linoleic acid, the major omega-6 PUFA, have doubled.56, 60 It is likely that all these factors have conspired to produce the current epidemic.
Each of the experimental diets often used to induce fatty liver in laboratory animals—diets high in fat, high in fructose, or deficient in choline and methionine—fail to completely capture the picture of human fatty liver disease. When these factors are combined, however, the picture begins to emerge (see sidebar).
The requirement for choline and the propensity to develop fatty liver is influenced by a person’s genetics and background diet. Diets rich in fat or fructose will require more choline than diets rich in starch. Vitamin B6, folate, vitamin B12 and betaine (a nutrient found most abundantly in spinach and to a lesser extent in wheat and beets) all reduce the requirement for choline. A person’s ability to make choline out of the amino acid methionine depends on his genetics, and preliminary studies suggest that Asians are better able to make this conversion than Caucasians.87-91
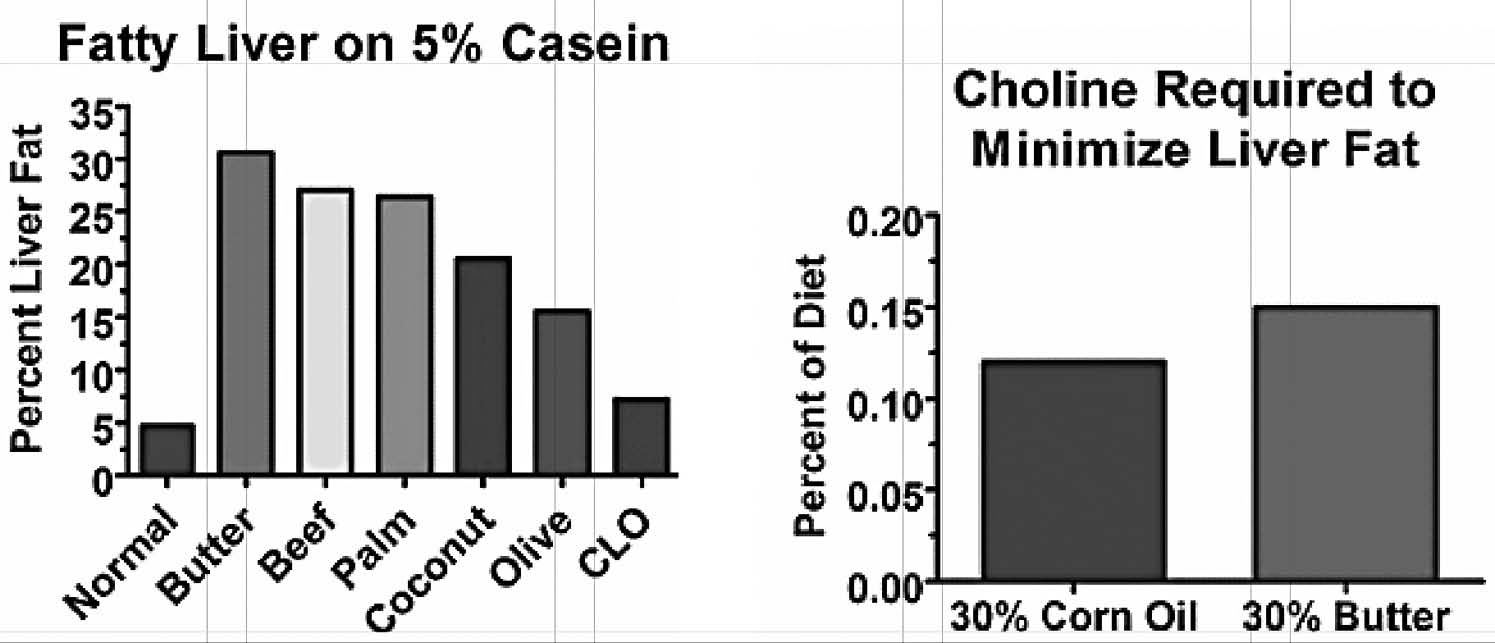
Figure 2. Left: Predominantly unsaturated or medium-chained fats produced less steatosis than predominantly long-chain saturated fats. Right: A diet containing 30 percent butter produced a choline requirement that was 30 percent higher than a diet containing 30 percent corn oil when fed to rats. From references 81 and 85.
Many other nutritional, metabolic, and lifestyle factors are likely to play a role in fatty liver as well, by influencing the liver’s ability to store carbohydrates as glycogen and to burn carbohydrates and fats for fuel. Thus, while there are special roles for including egg yolks, liver, other organ meats, and spinach in the diet, as well as avoiding polyunsaturated oils and refined foods—especially sugar—there is likely to be a wide range of different diets that can promote liver health. What they all have in common is that they are ancestral diets, rich in nutrient-dense foods that we are well-adapted to, rather than the displacing foods of modern commerce. The emergence of fatty liver as a silent epidemic in the modern era is a call to nourish our livers with age-old traditional wisdom as we pursue the vibrant health of our ancestors.
TABLE 1. Choline in Selected Foods
Data taken from the USDA Database for Choline Content of Common Foods. http://www.nal.usda.gov/fnic/foodcomp/Data/Choline/Choline.pdf
| Food | Choline (mg/100 g) | Food | Choline (mg/100 g) |
| Egg yolk, raw | 682 | Pistachios | 72 |
| Beef liver, pan-fried | 418 | Chicken, roasted | 66 |
| Chicken liver, pan-fried | 309 | Salmon, dry heat | 65 |
| Whole egg, raw | 251 | Cashews | 61 |
| Turkey Heart, simmered | 173 | Pine nuts | 56 |
| Wheat germ | 152 | Almonds | 52 |
| Bacon, pan-fried | 131 | Macadamia nuts | 45 |
| Mutton, roasted | 100 | Brussels sprouts, boiled | 41 |
| Turkey gizzard, simmered | 82 | Pecans | 41 |
| Shrimp, canned | 81 | Broccoli, boiled | 40 |
| Hamburger, broiled | 81 | Cauliflower, boiled | 39 |
References
1. Angulo P. Obesity and nonalcoholic fatty liver disease. Nutr Rev. Jun 2007;65(6 Pt 2):S57-63.
2. Wong VW, Wong GL, Choi PC, et al. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. Jul 2010;59(7):969-974.
3. Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. Jan 1990;11(1):74-80.
4. Lee RG. Nonalcoholic steatohepatitis: a study of 49 patients. Hum Pathol. Jun 1989;20(6):594-598.
5. Hamaguchi M, Kojima T, Takeda N, et al. Nonalcoholic fatty liver disease is a novel predictor of cardiovascular disease. World J Gastroenterol. Mar 14 2007;13(10):1579-1584.
6. Targher G, Bertolini L, Padovani R, et al. Prevalence of non-alcoholic fatty liver disease and its association with cardiovascular disease in patients with type 1 diabetes. J Hepatol. Oct 2010;53(4):713-718.
7. Zelman S. The liver in obesity. AMA Arch Intern Med. Aug 1952;90(2):141-156.
8. Brobeck JR, Tepperman J, Long CNH. Experimental Hypothalamic Hyperphagia in the Albino Rat. Yale J Biol Med. 1943;15(6):831-853.
9. Fenton PF, Chase HB. Effect of Diet on Obesity of Yellow Mice in Inbred Lines. Proc Soc Exp Biol Med. 1951;77(420-2).
10. Rabinowitch IM. Relationship Between Impairment of Liver Function and Premature Development of Arteriosclerosis in Diabetes Mellitus. Can Med Assoc J. 1948;58(6):547-556.
11. Root HF. Protamine Insulin in the Treatment of Diabetes Mellitus. Trans Am Clin Climatol Assoc. 1936;52:40-51.
12. Best CH, Hartroft WS, Lucas CC, Ridout JH. Liver Damage Produced by Feeding Alcohol or Sugar and Its Prevention by Choline. Br Med J. 1949;2(4635):1001-1006.
13. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. Jul 1980;55(7):434-438.
14. Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. Oct 2006;118(4):1388-1393.
15. Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology. Nov 1990;12(5):1106-1110.
16. Bellentani S, Saccoccio G, Masutti F, et al. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. Jan 18 2000;132(2):112-117.
17. Nomura H, Kashiwagi S, Hayashi J, Kajiyama W, Tani S, Goto M. Prevalence of fatty liver in a general population of Okinawa, Japan. Jpn J Med. May 1988;27(2):142-149.
18. Leite NC, Salles GF, Araujo AL, Villela-Nogueira CA, Cardoso CR. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. Jan 2009;29(1):113-119.
19. Prashanth M, Ganesh HK, Vima MV, et al. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. J Assoc Physicians India. Mar 2009;57:205-210.
20. Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, et al. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. Jul 2002;87(7):3023- 3028.
21. Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. Dec 2004;40(6):1387-1395.
22. Cowie CC, Rust KF, Ford ES, et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988-1994 and 2005-2006. Diabetes Care. Feb 2009;32(2):287-294.
23. Ford ES, Li C, Zhao G, Tsai J. Trends in obesity and abdominal obesity among adults in the United States from 1999-2008. Int J Obes (Lond). Sep 7 2010.
24. Daniel S, Ben-Menachem T, Vasudevan G, Ma CK, Blumenkehl M. Prospective evaluation of unexplained chronic liver transaminase abnormalities in asymptomatic and symptomatic patients. Am J Gastroenterol. Oct 1999;94(10):3010-3014.
25. Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. Feb 2005;288(2):E462-468.
26. Fischer LM, da Costa KA, Kwock L, Galanko J, Zeisel SH. Dietary choline requirements of women: effects of estrogen and genetic variation. Am J Clin Nutr. Nov 2010;92(5):1113- 1119.
27. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. Apr 18 2002;346(16):1221-1231. 28. Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology. Dec 1995;22(6):1714-1719.
29. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. Apr 1998;114(4):842-845.
30. Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A. Mar 18 1997;94(6):2557-2562.
31. Chung MY, Yeung SF, Park HJ, Volek JS, Bruno RS. Dietary alpha- and gamma-tocopherol supplementation attenuates lipopolysaccharide-induced oxidative stress and inflammatoryrelated responses in an obese mouse model of nonalcoholic steatohepatitis. J Nutr Biochem. Feb 4 2010.
32. Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. Oct 15 1999;274(42):30028-30032.
33. Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during nonalcoholic fatty liver disease. Diabetes Metab. Nov 2003;29(5):478-485.
34. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. May 2005;115(5):1343-1351.
35. Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. Mar 2006;116(3):817-824.
36. Charlton M, Sreekumar R, Rasmussen D, Lindor K, Nair KS. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology. Apr 2002;35(4):898-904.
37. Lee GS, Yan JS, Ng RK, Kakar S, Maher JJ. Polyunsaturated fat in the methionine-cholinedeficient diet influences hepatic inflammation but not hepatocellular injury. J Lipid Res. Aug 2007;48(8):1885-1896.
38. Letteron P, Fromenty B, Terris B, Degott C, Pessayre D. Acute and chronic hepatic steatosis lead to in vivo lipid peroxidation in mice. J Hepatol. Feb 1996;24(2):200-208.
39. Caldwell SH, Swerdlow RH, Khan EM, et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. Sep 1999;31(3):430-434.
40. Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. Nov 3 1999;282(17):1659-1664.
41. Perez-Carreras M, Del Hoyo P, Martin MA, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. Oct 2003;38(4):999- 1007.
42. Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. Apr 2001;120(5):1183-1192.
43. Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. Jan 1998;27(1):128-133.
44. Weltman MD, Farrell GC, Liddle C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology. Dec 1996;111(6):1645-1653.
45. Romestaing C, Piquet MA, Bedu E, et al. Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr Metab (Lond). 2007;4:4.
46. Kirpich IA, Gobejishvili LN, Homme MB, et al. Integrated hepatic transcriptome and proteome analysis of mice with high-fat diet-induced nonalcoholic fatty liver disease. J Nutr Biochem. Mar 19 2010.
47. Zou Y, Li J, Lu C, et al. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. Aug 8 2006;79(11):1100-1107.
48. Deng QG, She H, Cheng JH, et al. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology. Oct 2005;42(4):905-914.
49. Baumgardner JN, Shankar K, Hennings L, Badger TM, Ronis MJ. A new model for nonalcoholic steatohepatitis in the rat utilizing total enteral nutrition to overfeed a highpolyunsaturated fat diet. Am J Physiol Gastrointest Liver Physiol. Jan 2008;294(1):G27-38.
50. Lieber CS, Leo MA, Mak KM, et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. Mar 2004;79(3):502-509.
51. Ronis MJ, Korourian S, Zipperman M, Hakkak R, Badger TM. Dietary saturated fat reduces alcoholic hepatotoxicity in rats by altering fatty acid metabolism and membrane composition. J Nutr. Apr 2004;134(4):904-912.
52. Rivera CA, Gaskin L, Allman M, et al. Toll-like receptor-2 deficiency enhances nonalcoholic steatohepatitis. BMC Gastroenterol. 2010;10:52.
53. You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. Sep 2005;42(3):568-577.
54. Nanji AA, Sadrzadeh SM, Yang EK, Fogt F, Meydani M, Dannenberg AJ. Dietary saturated fatty acids: a novel treatment for alcoholic liver disease. Gastroenterology. Aug 1995;109(2):547-554.
55. Stephen AM, Wald NJ. Trends in individual consumption of dietary fat in the United States, 1920-1984. Am J Clin Nutr. Sep 1990;52(3):457-469.
56. Kim SY, Breslow RA, Ahn J, Salem N, Jr. Alcohol consumption and fatty acid intakes in the 2001-2002 National Health and Nutrition Examination Survey. Alcohol Clin Exp Res. Aug 2007;31(8):1407-1414.
57. Cortez-Pinto H, Jesus L, Barros H, Lopes C, Moura MC, Camilo ME. How different is the dietary pattern in non-alcoholic steatohepatitis patients? Clin Nutr. Oct 2006;25(5):816-823.
58. Ackerman Z, Oron-Herman M, Grozovski M, et al. Fructose- induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension. May 2005;45(5):1012-1018.
59. Sanchez-Lozada LG, Mu W, Roncal C, et al. Comparison of free fructose and glucose to sucrose in the ability to cause fatty liver. Eur J Nutr. Feb 2010;49(1):1-9.
60. Bray GA, Nielsen SJ, Popkin BM. Consumption of highfructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. Apr 2004;79(4):537- 543.
61. Assy N, Nasser G, Kamayse I, et al. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can J Gastroenterol. Oct 2008;22(10):811-816.
62. Ouyang X, Cirillo P, Sautin Y, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. Jun 2008;48(6):993-999.
63. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. May 2008;49(5):1068-1076.
64. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. Jan 2004;40(1):47-51.
65. Pickens MK, Yan JS, Ng RK, et al. Dietary sucrose is essential to the development of liver injury in the methionine- choline-deficient model of steatohepatitis. J Lipid Res. Oct 2009;50(10):2072-2082.
66. Holman RT. George O. Burr and the discovery of essential fatty acids. J Nutr. May 1988;118(5):535-540.
67. Burr GO, Burr MM. A New Deficiency Disease Produced by the Rigid Exclusion of Fat from the Diet. J Biol Chem. 1929;82(2):345-367.
68. Burr GO, Burr MM. On the Nature and Role of the Fatty Acids Essential in Nutrition. J Biol Chem. 1930;86(2):587- 621.
69. Jackson CM. The Effects of High Sugar Diets on the Growth and Structure of the Rat. J Nutr. 1930;3:61-67.
70. Bieri JG, Stoewsand GS, Briggs GM, Phillips RW, Woodard JC, Knapka JJ. Report of the American Institute of Nutrition Ad Hoc Committee on Standards for Nutritional Studes. J Nutr. 1977;107:1340-1348.
71. Bieri JG. Second Report of the ad hoc Committee on Standards for Nutrition Studies. J Nutr. 1980;110:1726.
72. Bacon BR, Park CH, Fowell EM, McLaren CE. Hepatic steatosis in rats fed diets with varying concentrations of sucrose. Fundam Appl Toxicol. Oct 1984;4(5):819-826.
73. Reeves PG. Components of the AIN-93 diets as improvements in the AIN-76A diet. J Nutr. May 1997;127(5 Suppl):838S-841S.
74. Reeves PG, Nielsen FH, Fahey GC, Jr. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. Nov 1993;123(11):1939-1951.
75. Banting FG. Diabetes and Insulin. Nobel Lecture. 1923.
76. Hershey JM, Soskin S. Substitution of “lecithin” for raw pancreas in the diet of the depancreatized dog. Am Physiological Soc. 1931;98:74-85.
77. Kaplan A, Chaikoff IL. The Effect of Choline on the Lipid Metabolism of Blood and Liver in the Completely Depancreatized Dog Maintained With Insulin. J Biol Chem. 1937;120(2):647-657.
78. Montgomery ML, Entenman C, Chaikoff IL, Feinberg H. Anti-fatty liver activity of crystalline trypsin in insulin-treated depancreatized dogs. J Biol Chem. Jul 1950;185(1):307-310.
79. Best CH, Hershey JM, Huntsman ME. The effect of lecithine on fat deposition in the liver of the normal rat. J Physiol. 1932;75(1):56-66.
80. Best CH, Huntsman ME. The effects of the components of lecithine upon deposition of fat in the liver. J Physiol. 1932;75(4):405-412.
81. Channon HJ, Wilkinson H. The effect of various fats in the production of dietary fatty livers. Biochem J. 1936;30(6):1033-1039.
82. Channon HJ, Wilkinson H. Protein and the dietary production of fatty livers. Biochem J. 1935;29(2):350-356.
83. Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem. Feb 25 1988;263(6):2998- 3004.
84. Buchman AL, Ament ME, Sohel M, et al. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: proof of a human choline requirement: a placebo-controlled trial. JPEN J Parenter Enteral Nutr. Sep-Oct 2001;25(5):260-268.
85. Benton DA, Elvehjem CA, Harper AE, Quiros-Perez F, Spivey HE. Effect of different dietary fats on choline requirement of rats. Proc Soc Exp Biol Med. Jan 1957;94(1):100-103.
86. Pan M, Cederbaum AI, Zhang YL, Ginsberg HN, Williams KJ, Fisher EA. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J Clin Invest. May 2004;113(9):1277-1287.
87. da Costa KA, Kozyreva OG, Song J, Galanko JA, Fischer LM, Zeisel SH. Common genetic polymorphisms affect the human requirement for the nutrient choline. FASEB J. Jul 2006;20(9):1336-1344.
88. Dong H, Wang J, Li C, et al. The phosphatidylethanolamine N-methyltransferase gene V175M single nucleotide polymorphism confers the susceptibility to NASH in Japanese population. J Hepatol. May 2007;46(5):915-920.
89. Song J, da Costa KA, Fischer LM, et al. Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD). FASEB J. Aug 2005;19(10):1266-1271.
90. Xu X, Gammon MD, Zeisel SH, et al. Choline metabolism and risk of breast cancer in a population-based study. FASEB J. Jun 2008;22(6):2045-2052.
91. Zhou YJ, Li YY, Nie YQ, et al. Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese people. J Gastroenterol Hepatol. Apr 2010;25(4):772-777.
This article appeared in Wise Traditions in Food, Farming and the Healing Arts, the quarterly journal of the Weston A. Price Foundation, Spring 2011.
🖨️ Print post
High Fat Diet
It seems you are saying a diet rich in animal fats (lard) contributes to Steatosis? So should we be concentrating on only butter and coconut oil for our main sources of fat in our diets because I like to add Suet to my grass-fed steaks.
Also, have you done much research into the liver’s connection with the lymphatic system and how longer chain fats are dumped directly into the lymphatic system opposed to other foods which have to pass through the liver first?
This was a long winded way of saying that Fructose = Metabolic Syndrome and Fatty Liver disease.
I’ve recently been diagnosed with a fatty liver. I no longer have a gallbladder (removed 10 yrs ago). They feel this has caused the fatty liver, I feel it’s been a very poor diet, (yo yo dieting). Based on the information provided it would be helpful to know how much of the Chlorine I would need to try to correct this liver problem. Can you please help with this.
Thank you, Kim
OMG It is Choline – not Chlorine!
Fatty liver
Just to clearify what you are saying. I have been told I have a fatty liver. I would like to reverse this a quickly and safely as possible. I had my gallbladder removed over 10 years ago and they feel this has contributed to the fatty liver. I feel it has contributed, possibly, but I feel my horrible diet over the years is the main reason. Can you tell me how much of the foods listed above I should consume daily to help with healing my liver.
Any help would be greatly appreciated. Kim Karas
NASH
I have been diagnosed with NASH and the Hepathologist recommended losing weight and 800 IU’s of Vitamin E to treat NASH. Sandy Cohen
Might the mucin accumulation of hypothyroidism be the cause of fatty liver?
Broda Barnes has a great deal of suggestive evidence that points to this.
Choline Supplements
“would be helpful to know how much of the Chlorine I would need to try to correct this liver problem.”
From my reading, about 1 gram/day (three to four caplets) appears to be sufficient. The stuff is pretty cheap at GNC and Vitamin Shoppe.
http://www.vitaminshoppe.com/s…id=VS-1166
http://www.gnc.com/product/ind…Id=2133450
question
plz tell me the amount of choline i should take per day to remove fat from my fatty liver its now four month since i was diognised with fatty liver but it has not reversed yet
Chris,
A recent study found that men with low testosterone have a greatly increased risk of NAFLD. The relative risk of men in the lowest quintile of T was 4.5 to five times greater, depending on how many things were adjusted for.
http://www.ncbi.nlm.nih.gov/pubmed?term=22691278
Poor diet, of course, could be a cause of low testosterone. Several studies have found that increased intake of vitamin K2, but not K1, significantly increases testosterone in rats.
On a different subject, I am baffled about how to post a comment on the Daily Lipid blog. I have tried and failed a number of times.
The role of PUFA, and especially linoleic acid, in NASH is clarified by the fact that LA is preferentially converted to cholesterol (22%) and other lipids in the liver (the same rate of conversion is conserved in rodent liver at very low intakes).
The concomitant effect of LA, upregulating of LDL-receptors, means that additional cholesterol is being brought into the liver at the same time. This effect overrides the usual controls on HMG-CoA reductase, so that dietary cholesterol can also contribute to overload.
Because cholesterol is forming at a rate that may exceed the liver’s ability to neutralise it by conjugation or esterification, and fatty acids may be formed faster than they can be neutralised as triglycerides, this results in a preponderance of free or unesterified cholesterol and other free lipids, which are far more reactive than neutral lipids, and can trigger oxidation and apoptosis in hepatic mitochondria.
I lost 140 pounds and I have a very strict diet I eat organic , no wheat , no bread , no pasta , no rice , no alcohol, no pop , no dairy , no coffee , no restaurant junk food , Carbs turn to sugar and your become insulin resistant and you end up with a fatty liver and gaining weight I eat mostly vegetarian but eat some fish and organic fried liver with coconut oil , and I jog 1 hour every day . also sleeping is very important if you have sleep apnea you will not lose weight unless you go for gastric surgery and follow a strict diet ! for the rest of your life ! carbs and sugar cause heart disease , fatty liver , cancer , weight gain .
l- Taurine is a good supplement to take for fatty liver and also drink filter water room tempeture with squeeze lemon every day ! the liver likes warm water not cold .
Milk thistle is something my husband has successfully used from time to time, to get better liver values. it normalizes the values. It’s believed to be protective of the liver. He only uses it short term since he tends to get stomach upset from it. He takes multivitamins daily, just can’t take milk thistle so often.
Yes, but it can be a problem for people with a ragweed allergy ad it is in the same family. I uded to take it occasionally but began getting hives.
A recent study found that men with low testosterone have a greatly increased risk of NAFLD. The relative risk of men in the lowest quintile of T was 4.5 to five times greater, depending on how many things were adjusted for.
http://saynotodisease.com/
Poor diet, of course, could be a cause of low testosterone. Several studies have found that increased intake of vitamin K2, but not K1, significantly increases testosterone in rats.
On a different subject, I am baffled about how to post a comment on the Daily Lipid blog. I have tried and failed a number of times.
Why doesn’t anyone talk about the real reason for liver problems? Alcohol!
What is alcohol but an extremely processed food? The alcoholics who succeed in getting sober tend to shift their addiction to cookies, candies and other processed foods. (From a voice of experience.)
Liver damage happens due to enormous intake of alcohol also.
What about avocado oil?