





Translations: Spanish | French
Article Summary
• Between 1928 and 1945, Weston Price measured the fat-soluble vitamin content of over twenty thousand samples of butterfat from many different regions. He found that abundant sunshine and rainfall, together with high-quality soil, was associated with high concentrations of vitamins within the butter and fewer deaths from heart disease.
• Modern science has shown that vitamins A, D, and K cooperate to prevent the calcification of arterial plaque, which in turn prevents heart disease. This confirms Price’s conclusions that fat-soluble vitamins protect against heart disease.
• We can maximize our fat-soluble vitamin status by consuming a diet rich in organ meats, animal fats, fatty fish, cod liver oil, and fermented foods, supplemented with leafy greens and other colorful vegetables; by spending lots of time out in the fresh air and sunshine; and by using traditional fats and oils while avoiding modern vegetable oils.
• Vitamin D can be a double-edged sword: adequate vitamin D prevents heart disease, but too much vitamin D promotes heart disease. The available evidence suggests that the lowest risk of heart disease occurs when vitamin D status is between 20 and 40 ng/mL.
• Trying to determine optimal vitamin D status is very problematic. Rather than trying to achieve an optimal vitamin D status with vitamin D supplementation, most people should focus more on optimizing the nutrient density and nutrient balance of the diet.
Fat-Soluble Vitamins in the Prevention of Heart Disease
In the second edition of Nutrition and Physical Degeneration, Weston Price published data suggesting that fat-soluble vitamins from animal fats might protect against heart disease. Between 1928 and 1945, Price collected over twenty thousand samples of butterfat to analyze for fat-soluble vitamins, from many regions across the United States, northwestern Canada, Australia, Brazil and New Zealand.
His data suggested that abundant sunshine, rainfall and high-quality soil led to an abundance of rapidly growing, lush, richly green grass, butterfat rich in fat-soluble vitamins, and fewer deaths from heart disease. Our understanding of heart disease has progressed immensely since Price’s time, and now more than ever we can be confident that Price’s emphasis on the protective power of fat-soluble vitamins was correct.
Food Sources of the Fat-Soluble Vitamins
Price placed special emphasis on vitamins A, D, and K. These vitamins are richest in butterfat when cattle are raised in the open sunshine and consume richly green grass, leading to a deeply yellow or even orange butterfat. Price also noted other important sources of fat-soluble vitamins. He concluded from his studies of traditional peoples that while some groups obtained fat-soluble vitamins primarily from dairy foods, others obtained them primarily from organ meats and eggs, from the animal life of the sea, or from insects and other small animals. In his own practice, he emphasized organ meats, cod liver oil, butterfat and fish, supplemented by colorful vegetables, as sources of fat-soluble vitamins. What we know about food sources of fat-soluble vitamins in our own era could be summarized as follows.
Vitamin A is found only in animal foods. Animals store vitamin A primarily in their livers. As a result, the best sources of vitamin A are the livers of land animals or fish. Oil extracts of these livers, such as cod liver oil, are similarly excellent sources of vitamin A. Although not often used as food in modern diets, eyeballs contain even higher concentrations of the vitamin because of its important role in vision, as does the tissue located behind the eyeballs.
Smaller amounts of vitamin A are found in the fatty tissue of animals. Because of its critical role in growth and development, the fats most closely related to reproduction—butterfat, meant to nourish a young animal, and egg yolks, meant to become a young animal—tend to be richer in vitamin A than other animal fats.
Carotenoids from plant foods are often confused with vitamin A, but they are not the same thing. There are over six hundred known carotenoids, roughly 10 percent of which are precursors to vitamin A. Among these, the most important in our diets are beta-carotene, alphacarotene and beta-cryptoxanthin. Carotenoids provide plants with red, orange and yellow colors. Because they play an important role in photosynthesis, they are closely associated with chlorophyll, which imparts a green color. Red, orange, yellow and green colors thus provide a strong indication that a plant is rich in carotenoids, which we can potentially convert to vitamin A.
Many factors affect the ability to convert carotenoids to vitamin A,1,2 making them a highly variable and less reliable source of the vitamin than animal foods. The percentage of carotenoids converted to vitamin A ranges from 3 to 25 percent for most plant foods. The conversion is much higher from foods with simple matrices; as a result, it is highest from red palm oil, intermediate for fruits, and lowest for vegetables. Cooking and puréeing fruits and vegetables, however, increases the conversion. Fiber, parasites, toxic metals, oxidative stress and deficiencies of iron, zinc, protein and thyroid hormone all decrease the conversion. Conversely, fat, vitamin E and a deficiency of vitamin A increase the conversion. It is quite easy to see how complex this issue can become. Someone who is deficient in vitamin A will make more of it from plant foods, but what if that person is also deficient in iron and protein, or suffers from hypothyroidism?
Even if all these factors are optimized, there is a strong effect of genetics. Almost half of people with European ancestry have a genetic mutation that decreases their ability to make the conversion at least twofold, and about a third have a second mutation that decreases their ability to make the conversion fourfold.3 Thus, while many people may be able to extract adequate vitamin A from plant foods, many may not. For the latter, even if they use red palm oil, cook or purée their fruits and vegetables, eat those fruits and vegetables with fat, minimize their exposure to toxins, and have healthy digestive systems and optimal hormonal status, their genetics will prevent them from satisfying their need for this vitamin from plants alone. The inclusion of colorful fruits and vegetables in the diet is an excellent way to supplement more reliable sources of vitamin A, but the inclusion of nutrient-dense animal foods in the diet is a critically important insurance policy against vitamin A deficiency and a more reliable and robust way of optimizing vitamin A status.
We obtain vitamin D through exposure to sunshine and from consuming the bodies of fatty fish, the livers and liver oils of fish, and in smaller amounts from other animal fats, especially butterfat and egg yolks.
Vitamin K comes in two forms: vitamin K1 and vitamin K2. Vitamin K1 is most abundant in leafy greens, while vitamin K2 is most abundant in animal fats and fermented foods. The richest sources of vitamin K2 in modern diets are egg yolks and cheese, especially hard cheeses. While much more data documenting the distribution of vitamin K2 in foods is needed, current databases suggest that the richest sources of the vitamin are natto, a fermented soy food common in Eastern Japan, and goose liver.
Vitamin K2 appears to be much more effective at preventing pathological calcification than vitamin K1, but there is some overlap between the two, and humans have a limited ability to convert K1 to K2. Emerging evidence also suggests that the form of vitamin K2 found in animal foods has unique functions not possessed by the form found in fermented foods. The wisest approach to vitamin K nutrition seems to be to cover all the bases by eating a diet rich in leafy greens, animal fats and fermented foods.
When looking at nutritional databases, it is important to keep in mind that these databases universally ignore the variation in nutrition between different foods. “Butter” is likely to have a single value for each nutrient, but a major point of Price’s analysis of over twenty thousand butter samples was the extreme variation in nutrient values. The factors responsible for this variation are discussed in detail in the sidebar below.
It is also important to keep in mind that the context in which these foods are eaten determines the availability of their nutrients. Fat, for example, is critical to the absorption of fatsoluble vitamins. The absorption of carotenoids from salad with no added fat is close to zero, while the addition of canola oil increases their absorption.4 The type of fat also matters. Compared to safflower oil, beef tallow promotes better absorption of beta-carotene and better conversion to vitamin A.5 Similarly, olive oil promotes better carotenoid absorption than corn oil.6 It appears from the available evidence that traditional fats and oils emphasizing saturated and monounsaturated fatty acids promote much better fat-soluble vitamin absorption when compared to modern polyunsaturated vegetable oils.
Overall, then, we can maximize our intake of these vitamins by consuming liver, cod liver oil, fatty fish, animal fats and fermented foods, and by getting plenty of fresh air and sunshine. Fruits and vegetables displaying red, orange, yellow, and green colors help supplement our intakes of these vitamins. Adding traditional fats and oils to the diet, while excluding modern vegetable oils, helps maximize the biological activity of these vitamins.
Protection Against Calcification and Plaque Rupture
While there are likely many ways this fat-soluble trio protects against heart disease, this article will focus on the most well established link: by protecting against the calcification of arteries, these vitamins in turn protect against the rupture of atherosclerotic plaques. Plaque rupture is the principal cause of the narrowing of coronary arteries and the formation of deadly clots in these arteries, and thus plays a major role in heart attack and stroke (see sidebar).
Until recently, most heart disease researchers considered the calcification of arterial plaque to be a phenomenon that starts only after atherosclerosis has become severe. They questioned, moreover, whether such calcification is truly harmful in and of itself or is simply a marker for the overall severity of the disease.
In the past few years, however, it has become clear that calcification begins in the very earliest stages of atherosclerosis.7 In hindsight, it is not too surprising that researchers previously missed this: nearly all of the calcification in a plaque site—a full of 97 percent of it, in fact—is so small that modern imaging equipment designed to visualize calcification in a live human being is incapable of detecting it.8 These “microcalcifications” make a plaque up to five times more likely to rupture under stress.8 Depending on the severity of the rupture, this will lead either to greater narrowing of the artery or to a cardiovascular “event” such as a heart attack or stroke (see sidebar).
The calcification of atherosclerotic plaque occurs in parallel with the accumulation of a defective, inactive form of matrix Gla protein (MGP).7 This fact provides a strong hint about the role of the fat-soluble trio: as has been known for quite some time, vitamins A and D cooperate together to control how much MGP our cells produce; once produced, vitamin K activates the protein, thereby enabling it to control the distribution of calcium.9
Indeed, mice with a genetic defect preventing them from producing MGP fail to accumulate calcium in their bones, suffer from osteopenia and spontaneous fractures, and yet die within two months of birth from the rupture of heavily calcified arteries.10 It is primarily vitamin K2, found in animal fats and fermented foods, that activates MGP, which likely explains why people with the highest intakes of vitamin K2—primarily from egg yolks and cheese—have much lower rates of arterial calcification and coronary heart disease.11
Vitamin D: A Double-Edged Sword?
While vitamin K, especially vitamin K2, seems to be straightforwardly protective, the story of vitamins A and D is more complex. When vitamins A and D are both provided abundantly, they maximize the protective effect of vitamin K, but when vitamin D is provided in great excess of vitamin A, it actually promotes abnormal, pathological calcification of soft tissues, including arteries.12,13,14 This finding suggests that vitamin D may be a double-edged sword, with the ability to either prevent or promote heart disease, depending on the dietary context in which it is provided.
Indeed, both severe deficiencies of vitamin D15 and hefty excesses of the vitamin16 promote atherosclerosis in animal experiments. Observational studies in humans show that the risk of heart disease declines as vitamin D status increases. This relationship plateaus at about 24 ng/mL, and there is very little data exploring higher levels (see Figure 1). However, a recently published study suggested that having vitamin D status higher than 40 ng/mL is just as dangerous as having vitamin D status lower than 12 ng/ mL (see Figure 2). When viewed together, the evidence in animals and humans suggests that vitamin D protects against heart disease at the right dose, but that too much vitamin D actually contributes to heart disease.
![]()
Figure 1: The Risk of Cardiovascular Disease Declines with Increasing 25(OH)D Up to 24 ng/mL
This figure is adapted from Figure 3 as originally published in reference 27. The horizontal axis has been converted from nmol/L to ng/mL so that the units correspond to those used by clinical laboratories in the United States. The figure depicts data pooled from sixteen independent studies measuring serum 25(OH)D and subsequent risk of cardiovascular disease. 25(OH) D is a metabolic product of vitamin D that is often used as a measure of vitamin D status, though there are problems with this. Each circle represents an independent risk estimate for a given category of 25(OH)D from an individual study. The size of the circle represents the statistical power of the study, driven in part by low variation but mostly by large sample size. Circles further to the right represent higher concentrations of 25(OH)D and those higher up represent higher risks of cardiovascular disease. The shaded area represents the confidence interval. The more narrow the shaded area, the higher our confidence in the estimates; the wider the shaded area, the more uncertainty we have.
The risk of cardiovascular disease declines with increasing 25(OH)D up to 24 ng/mL but appears to plateau thereafter. There are only two data points with poor statistical power at concentrations higher than 32 ng/mL and there are no data points at concentrations higher than about 45 ng/mL. The paucity of the data in these regions makes the uncertainty surrounding the risk estimate very high, represented by the increasingly wide shaded area.
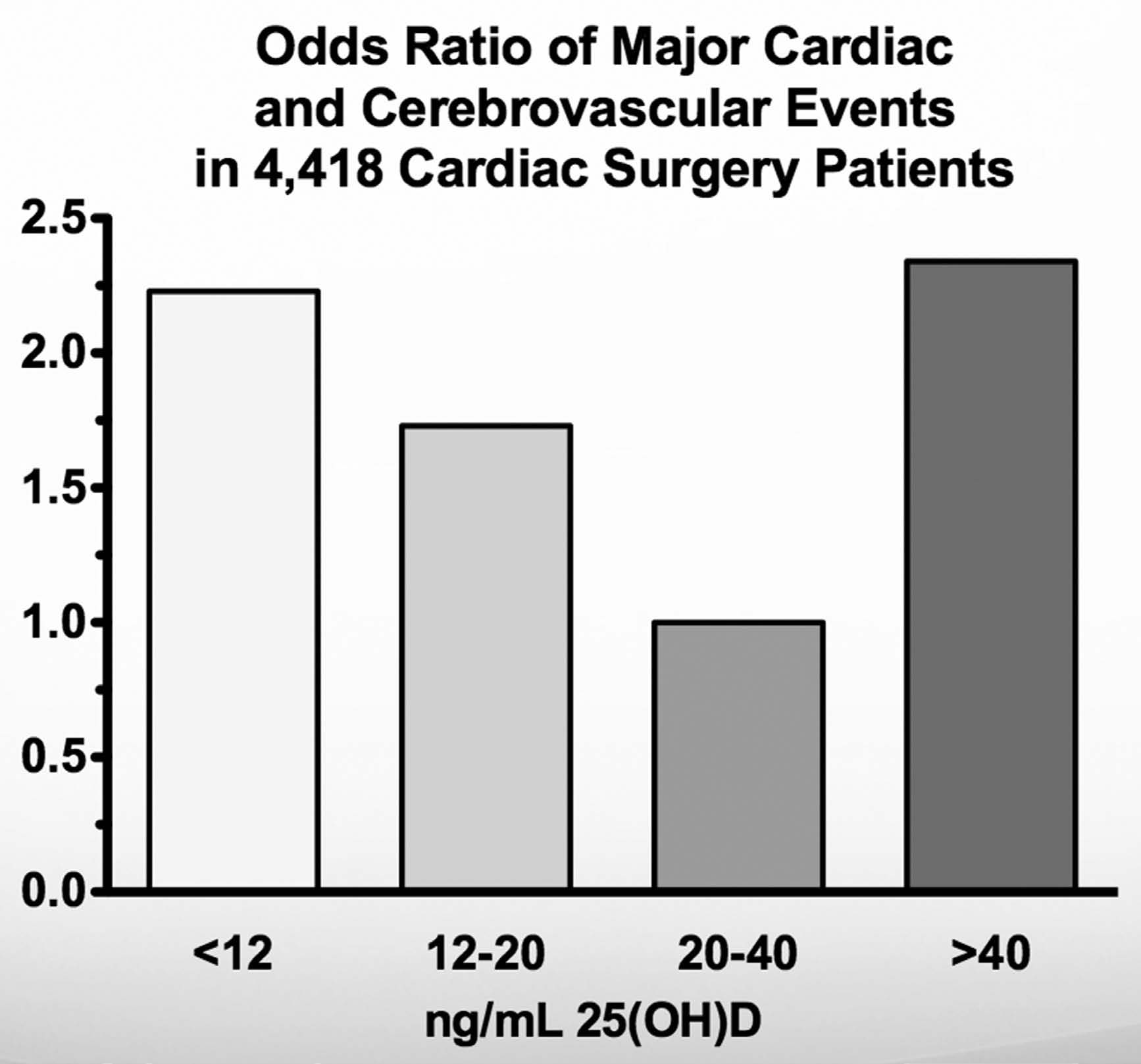
Figure 2: The Risk of Cardiovascular Events is Lowest between 20-40 ng/mL among Cardiac Surgery Patients
This figure is adapted from the data in reference 28. The researchers measured serum 25(OH)D in just under 4,500 cardiac surgery patients, in whom the risk of future cardiovascular events was very high. Over the following year, 11.5 percent of the patients suffered a major event. The risk decreased with increasing concentrations of 25(OH)D up to 40 ng/mL, but increased thereafter. Those with 20-40 ng/mL had the lowest risk, but those with concentrations greater than 40 ng/mL had just as high a risk as those with less than 12 ng/mL.
Many readers may be surprised that people with vitamin D status higher than 40 ng/mL have a higher risk of heart disease when so many advocates of vitamin D supplementation recommend levels much higher than this. Part of the reason many people recommend higher levels is because they view the evidence within the framework of the very influential but very problematic “naked ape” hypothesis of optimal vitamin D status (see sidebar).
We should keep in mind, however, that none of these studies takes into account the interactions between vitamins A, D and K. It may be that vitamin D status higher than 40 ng/mL protects against heart disease in the context of a diet that provides liberal amounts of organ meats, animal fats and fermented foods. It may also be that a simple cause-and-effect relationship between vitamin D exposure and serum 25(OH)D, or between serum 25(OH)D and disease risk, greatly oversimplifies the issue (see sidebar). The uncertainty over these questions underlines the need to pay more attention to optimizing the nutrient density and nutrient balance of the diet rather than overemphasizing the usefulness and importance of optimizing blood levels of vitamin D.
An Old Solution to a New Problem
The successful traditionally living groups that Price studied placed special emphasis on procuring foods rich in fat-soluble vitamins, supporting the health of their animals, and taking great care to preserve the health of their soil. The causes of the twentieth century emergence of heart disease are debatable, but Price’s suggestion that the fat-soluble vitamins provide powerful protection against the disease has gained validation through decades of further scientific inquiry. There is little doubt that the emergence of refined foods, the replacement of butter with substitutes based on vegetable oils, the demonization of eggs, the loss of traditions centered on the use of liver and cod liver oil, the dilution of the nutritional value of animal products through industrial farming, and the campaign against animal fats have all greatly diminished our ability to prevent and reverse this disease. The pervasive view that the foods richest in fat-soluble vitamins are the very causes of heart disease because of their saturated fat and cholesterol is particularly ironic and especially harmful. Returning to the traditional emphasis on foods rich in fat-soluble vitamins may not be the whole answer but it is a critical piece of the puzzle and an essential tool in our kit as we work toward a world where we prevent the inevitable and cure the incurable.
Sidebars
Price’s Analysis of Butter Samples: A Closer Look
Price collected samples of butterfat every two to four weeks from many different regions. This allowed him to trace the fat-soluble vitamin content of the butter through the year in each region. He also assembled data provided by other researchers showing how sunshine, rainfall and mortality from heart disease and pneumonia varied through the year in the same regions.
Price presented the mortality data for both diseases combined. This makes the graphs cleaner and more readable but precludes us from analyzing the trends for heart disease and pneumonia separately. In all likelihood, the fat-soluble vitamins found in the butterfat protected against both diseases. Clinical trials over the preceding two decades had clearly demonstrated the power of vitamins A and D—and cod liver oil, which contains a rich supply of both vitamins—to protect against many different infectious diseases.17,18 The main text of this article focuses on the evidence supporting the power of these vitamins and their synergistic partner, vitamin K, to protect against heart disease.
Although this article focuses on vitamins A, D, and K as a synergistic trio, Price only used two chemical tests to look for fat-soluble vitamins. His test for vitamin A used toxic reagents and lacked perfect specificity—it picked up carotenoids, for example, which are also present in butter—but it was a good test, dominant through the 1970s, and is still used in some laboratories today. Price’s second test, however, has a more complicated story behind it.
From Price’s time through our own, scientists have primarily used the test to detect lipid peroxides, which are formed when delicate polyunsaturated fatty acids suffer oxidative damage. Based on research suggesting a correlation between an oil’s potential to oxidize and its vitamin D content, Price initially used the test as an imperfect way to measure vitamin D. It soon became clear, however, that isolated vitamin D caused soft tissue calcification. Butterfat scoring high on the test, by contrast, seemed to safely and effectively promote the calcification of bones and teeth, especially when combined with cod liver oil, and seemed to have broader activities that no one had yet ascribed to vitamin D. Price therefore dropped the term “vitamin D” from his butterfat analysis and began using the term “activator X.”
Researchers publishing in Russian- and German-language literature, unbeknownst to Price and many others writing in English, had been using the same test to detect the synthetic chemical benzoquinone, which belongs to a class of chemicals known as quinones. Decades later, researchers publishing in English showed that the test detects biological quinones such as coenzyme Q10. Vitamin K is another such quinone, and appears to be the compound Price was trying to measure. It exists in two forms: K1 and K2. Cows obtain vitamin K1 from grass and convert a portion of it to vitamin K2. Both forms of the vitamin are present in the butterfat and presumably registered as “activator X” in Price’s test. Vitamin K2, moreover, has all the biological characteristics Price attributed to activator X. A comprehensive argument identifying activator X as vitamin K2 can be found in my Spring 2007 Wise Traditions article, “On the Trail of the Elusive X-Factor: A Sixty-Two-Year-Old Mystery Finally Solved.”9
Price wrote in Nutrition and Physical Degeneration that the vitamin A and activator X content of butterfat was related more to rainfall than sunshine, depended most closely on the rapid growth of lush, green grass, and peaked at much greater concentrations in regions where the soil remained most intact. There is a simple explanation for these findings. When grass rapidly grows, it ramps up its photosynthetic activity. Photosynthesis uses energy from sunlight and electrons from water to convert carbon dioxide to sugar, which is needed to fuel growth. Essential components of the photosynthetic machinery include chlorophyll, beta-carotene and vitamin K1.19 Chlorophyll imparts a deep green color to the grass. Cattle convert a portion of beta-carotene to vitamin A and a portion of vitamin K1 to vitamin K2. All four nutrients are present in the butterfat, and the beta-carotene imparts a yellow or even deeply orange color to it.
The photosynthetic machinery also depends critically on minerals from the soil, including iron, sulfur, calcium and magnesium.19 A deficiency of any other essential soil mineral can also limit the ability of a plant to ramp up photosynthesis. Boron, for example, is not directly involved in the photosynthetic machinery, but its deficiency compromises photosynthesis and depletes chlorophyll and beta-carotene.20 Lack of available boron could be caused by lack of boron itself or by factors that compromise its bioavailability such as high soil pH or low concentrations of organic matter. We can conclude from all this that the recipe for high vitamin A and activator X concentrations in butterfat is adequate water, sunshine and soil health, which together support the rapid growth of grass.
If Price was correct that the difference in peak vitamin content between different regions was primarily a result of soil quality, then it is clear from his data that soil health is often the limiting factor: regions with long histories of soil depletion had low levels of fat-soluble vitamins year-round. Price dismissed the role of sunshine and suggested rainfall was more dominant, probably because peak rainfall tends to occur when sunshine is already adequate and rainfall is more often the weakest of the two links in the chain. Had Price been able to resolve the conundrum presented by the “activator X” test, however, he would likely have recognized a greater role for sunshine. Cattle obtain vitamin D from the sun, not from grass. Price did not have access to a reliable chemical test for vitamin D, but other researchers at the time tested the vitamin D content of butterfat by measuring its ability to prevent rickets in experimental animals. These studies showed that the vitamin D content of butter correlates closely with the exposure of the cattle to sunshine.21 Indeed, if we analyze Price’s data closely, it seems that both sunshine and the vitamin content of butterfat are associated with fewer deaths from heart disease and pneumonia. This is probably because during periods of greater sun exposure, people made more of their own vitamin D and obtained more vitamin D from butterfat; when the grass was growing most rapidly, the butterfat also provided abundant amounts of vitamins A and K, enabling maximal synergy between the three vitamins.
Plaque Rupture and Coronary Heart Disease
An up-to-date review of the best evidence available today suggests that the current view of heart attacks—that most are caused by the occlusion of coronary arteries by blood clots known as thrombi or less often by severe narrowing of the coronary arteries—is largely correct.
The role of thrombi in heart attacks was highly controversial during the mid-twentieth century through the 1970s.22 Some studies found coronary thrombi in fewer than 10 percent of cases, while others found them in over 90 percent of cases. Some research even showed that clots were more often found when people had died at least twenty-four hours after the onset of a heart attack and were rarely found when they died within an hour of the heart attack, suggesting that clots may be a consequence rather than a cause of heart attacks, forming in the distant aftermath of a fatal event. Researchers tried to determine whether clots form before or after heart attacks by injecting people with radiolabeled fibrin soon after a heart attack to see whether the clots contained the radiolabel, but the results were conflicting and difficult to interpret.
In 1979, a team of English researchers made a compelling argument that several methodological and interpretive problems were at the root of the controversy.22 Even within the same hospital, some analysts were much more likely than others to find thrombi after heart attacks, largely because of variations in the methods used. Similarly, studies using more careful methods were more likely to find thrombi. Sudden death was often lumped together with heart attacks, even though there is no way to know that a sudden death is actually due to a heart attack. During a true heart attack, cells of the heart die and spill out certain enzymes into the blood. If the person lives, a doctor can verify a heart attack by finding these enzymes in the blood. If the person dies, a doctor can verify a heart attack by finding these enzymes missing from the heart tissue. To run this test at autopsy, however, the person must have died at least six hours after the heart attack, and the most accurate results are found when the person died at least twelve to twenty-four hours after. Thus, not finding coronary thrombi in someone who died within an hour of a heart attack could just be an indication that the person did not die of a heart attack at all. Most studies of the time didn’t adequately differentiate between different types of heart attacks. Heart attacks where the cell death is spread diffusely through the heart tissue are rarely associated with coronary thrombi but are frequently associated with severe narrowing of all three coronary arteries. Heart attacks where the cell death afflicts a very specific region of the heart are much more common and are almost always associated with coronary thrombi. Finally, the most carefully conducted and analyzed studies using radiolabeled fibrin suggested that coronary thrombi begin to form before a heart attack occurs, and after the heart attack they continue to grow and then eventually dissolve.
In 1980, angiography allowed researchers to look for the first time for coronary thrombi in live people suffering from heart attacks.23 Coronary thrombi were almost always present in the first six hours after the onset of symptoms. Between six and twenty-four hours after the onset of symptoms, thrombi were found less often, being present in 80-85 percent of cases and completely occluding a coronary artery in 65-70 percent of cases. This study contradicted the suggestion of earlier research that clots are more likely to form twenty-four hours after a heart attack occurs. Instead, it suggested that clots are almost universally present in the earliest hours and begin dissolving after six hours. This strengthened the alternative interpretation of earlier research, which held that thrombi were not found when people died a short time after their putative heart attack because they had suffered from misclassified cases of sudden death and in fact had not suffered from a heart attack at all.
Taken together, the evidence suggests that coronary thrombi are responsible for the more common regional heart attacks and that severe narrowing of the coronary arteries contributes to the less common diffuse heart attacks. Both of these factors reduce the supply of blood and the oxygen it carries, making cells vulnerable to death. None of this suggests that other factors are not important. Indeed, there may be many factors that make some cells vulnerable to damage during transient deprivation of oxygen and make others less vulnerable. There may also be other acute events involved that transiently deprive cells of oxygen or otherwise impair their metabolism, particularly in the less common diffuse heart attacks.
The principal cause both of coronary thrombi and coronary narrowing is plaque rupture. In the case of thrombi, rupture allows the inflammatory contents of the plaque to spill out into the blood and cause the sudden formation of a clot.24 The case of narrowing may seem less intuitive. When atherosclerotic plaque accumulates, it does not grow inward into the blood vessel like grease clogging a pipe. It actually accumulates inside the blood vessel wall, pushing the wall outward, allowing an equivalent or even larger space for blood to flow within the vessel.25 When the plaque environment becomes sufficiently inflammatory, plaque rupture ensues. If the aftermath of the rupture is mild, the plaque will heal itself. This healing process results in successive plaques overlaying each other, with each healed rupture intruding more and more into the inside of the artery.26 Thus, severely inflammatory ruptures contribute to an occlusive thrombus that may result in an immediate heart attack, while mild ruptures lead to progressive narrowing of the arteries, which impedes blood flow and could eventually contribute to a heart attack.
What causes plaques to rupture? As plaque develops, it forms a highly protective fibrous cap that is rich in collagen. The accumulation of oxidized lipids leads to an inflammatory environment that degrades collagen and prevents its synthesis.24 Lack of nutrients needed to synthesize collagen, such as vitamin C and copper, could play a role, as could infiltration of the plaque by infectious microbes. Plaques that are richest in oxidized lipids and poorest in collagen are most likely to rupture. As discussed in the main text, though, even when these factors are held constant, small deposits of calcium in the fibrous cap greatly increase its vulnerability to stress and make it far more likely to rupture. The fat-soluble trio—vitamins A, D and K—forms our principal defense against this calcification.
Problems With the “Naked Ape” Hypothesis of Optimal Serum 25(OH)D Concentrations
One of the most widely influential perspectives about 25(OH )D that appears in the scientific literature and pervades the alternative health literature is the “naked ape” hypothesis of optimal serum 25(OH )D. This hypothesis holds that humans evolved as “naked apes” in the tropical savannahs of Africa where they were exposed to maximal sunshine and when the requirement for 25(OH )D was indelibly fixed into our genome. Now that we have invented modern clothing, indoor living, and migrated far from the tropics, most of us have far lower 25(OH )D than we had “back when we evolved,” as shown by the much higher levels of 25(OH )D found in lifeguards working in Missouri and Israel. Reinhold Vieth promoted this view in a popular 1999 article.29 At the time of this writing, Google Scholar reports that this article has been cited 1,159 times.
While it may seem compelling on the surface, the argument is deeply problematic. The hypothesis assumes that at some point between the loss of body hair and the gain of clothing we existed as naked sunbathers, and it was at just this very point where the requirement for serum 25(OH )D was indelibly fixed into our genome. If we take “molecular clock” estimates at face value, the loss of body hair and the gain of dark skin pigmention both occurred 1.2 million years ago,30,31 indicating we were never truly “naked” since both hair and pigment protect the skin from ultraviolet light. Evidence for hide scrapers likely used to make leather, either for clothing or some other form of shelter from the sun such as housing, goes back almost eight hundred thousand years.30 Clothing was certainly in widespread use by the time clothing lice diverged from head lice, which scientists estimate occurred some one hundred seventy thousand years ago.30 Colored pigments appear in the African archeological record over a quarter million years ago and remain a constant feature of African culture through the present.32 These may have been used to paint the skin, as commonly occurs in Africa today. Weston Price wrote in Nutrition and Physical Degeneration that it was a universal tradition in the Pacific Islands to use coconut oil as a sunscreen, and there is no particular reason to doubt the premise that prehistoric humans used botanical sunscreens as well. African primates and traditionally living African humans seek shade from the hot sun at mid-day.33,34 Prehistoric humans living in the African savannah were thus likely to be neither “naked” nor sunbathers.
Most of prehistoric human life was dominated by glacial periods in which the earth was substantially colder and aerosolized dust and salt were much higher.35 The lower exposure of the earth to solar radiation and the higher aerosols during these times probably made the average UV-B exposure considerably lower, suggesting that no living human beings provide a proxy for prehistoric 25(OH )D levels. The worst example we could possibly use for such a purpose, however, are modern lifeguards. The Israeli lifeguards whose high 25(OH )D Vieth cited in his 1999 paper as the closest approximation to the vitamin D status of our “naked ape” ancestors had evidence of sun damage and twenty times the risk of kidney stones as the general population. The lifeguards had a mean 25(OH )D higher than 50 ng/mL, and their increased risk of soft tissue calcification is consistent with the increased risk of cardiovascular disease that occurs above 40 ng/mL (see Figure 2).
Additionally, certain populations seem adapted to a lower “normal” 25(OH )D. Greenland Inuit on their traditional diet have a mean serum 25(OH )D of only 20 ng/mL, but appear to convert 25(OH )D to the more active 1,25(OH )2D at a higher rate.36 Similarly, African Americans have lower 25(OH )D than white Americans, but higher 1,25(OH )2D and higher bone density.37 It seems that these groups have lower 25(OH )D but higher total biological activity of vitamin D, creating the illusion of a “deficiency” that does not actually exist. If different groups are adapted to different optimal levels of 25(OH )D, moreover, this suggests that the requirement for 25(OH )D has continued to evolve over time and was never indelibly fixed into the human genome at any point, certainly not in some fictitious era of the “naked ape.”
The very concept of an optimal 25(OH )D may itself be flawed. The total biological activity of vitamin D is determined by both 25(OH )D and the much more active 1,25(OH )2D. The conversion of 25(OH )D to 1,25(OH )2D is, like many other steps in vitamin D metabolism, partly determined by genetics.38 Many other factors can influence either the demand for or the supply of 1,25(OH )2D. Calcium deficiency increases the demand for it and lowers 25(OH )D status independently of vitamin D exposure.39 Vitamin A, by contrast, seems to increase the supply of 25(OH )D to the kidney, making it easier to convert it to 1,25(OH )2D.40 Acute inflammation41 and cancer42 also increase the conversion. If we only measure 25(OH )D and it is low, we have no idea whether total biological activity of vitamin D is increased or decreased, nor do we know why it is altered or whether this is a concern. The fact that crisis states such as acute inflammation and disease states such as cancer can influence the conversion raises an additional problem: are associations between 25(OH )D and disease risk cause or effect? Until these questions are resolved, we should place much less emphasis on using vitamin D supplements to achieve a desired 25(OH )D and much more emphasis on improving the nutrient density and nutrient balance of the diet.
REFERENCES
1. Haskell MJ. The challenge to reach nutritional adequacy for vitamin A: [beta]-carotene bioavailability and conversion – evidence in humans. Am J Clin Nutr. 2012;96(suppl):1193S- 203S.
2. Yamaguchi N, Suruga K. Triiodothyronine stimulates CMO1 gene expression in human intestinal Caco-2 BBd cells. Life Sci. 2008;82(13-14):789-96.
3. Leung WC, Hessel S, Meplan C, Flint J, Oberhauser V, Tourniaire F, Hesketh JE, von Lintig J, Lietz G. Two common single nucleotide polymorphisms in the gene encoding beta-carotene 15,15’-monooxygenase alter beta-carotene metabolism in female volunteers. FASEB J. 2009;23(4):1041-53.
4. Brown MJ, Ferruzzi MG, Nguyen ML, Cooper DA, Eldridge AL, Schwartz SJ, White WS. Carotenoid bioavailability is higher from salads ingested with full-fat than with fat-reduced salad dressings as measured with electrochemical detection. Am J Clin Nutr. 2004;80(2):396-403.
5. Hu X, Jandacek RJ, White WS. Intestinal absorption of [beta]-carotene ingested with a meal rich in sunflower oil or beef tallow: postprandial appearance I triacylglycerol- rich lipoproteins in women. Am J Clin Nutr. 2000;71:1170-80.
6. Clark RM, Yao L, She L, Furr HC. A comparison of lycopene and astaxanthin absorption from corn oil and olive oil emulsions. Lipids. 2000;35(7):803-6.
7. Roijers RB, Debernardi N, Cleutjens JP, Schurgers LJ, Mutsaers PH, van der Vusse GJ. Microcalcifications in early intimal lesions of atherosclerotic humans coronary arteries. Am J Pathol. 2011; 178(6):2879-87.
8. Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S. A mechanistic analysis of the role of micro calcifications in atherosclerotic plaque: stability potential implications for plaque rupture. Am J Physiol Heart Circ Physiol. 2012;303(5):H619-28.
9. Masterjohn C. On the Trail of the Elusive X Factor: A Sixty-Two-Year-Old Mystery Finally Solved. Wise Traditions. Spring, 2007.
10. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386(6620):78-81.
11. Geleijnse JM, Vermeer C, Grobbee DE, Schurgers LJ, Knapen MH, van der Meer IM, Hofman A, Witteman JC. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study. J Nutr. 2004;134(11):3100-5.
12. Masterjohn C. From Seafood to Sunshine: A New Understanding of Vitamin D Safety. Wise Traditions. Fall, 2006.
13. Masterjohn C. Vitamin D toxicity redefined: vitamin K and the molecular mechanism. Med Hypotheses. 2007;68(5):1026-34.
14. Masterjohn C. Thyroid Hormone and Vitamin A Protect Against Vitamin D Toxicity in Cows. Mother Nature Obeyed. Published April 3, 2013. https://www.westonaprice.org/our-blogs/thyroid-hormone-and-vitamin-a-protect-against-vitamin-d-toxicity-in-cows/ Accessed November 21, 2013.
15. Schmidt N, Brandsch C, Kuhne H, Thiele A, Hirche F, Stangle GI. Vitamin D receptor deficiency and low vitamin D diet stimulate aortic calcification and osteogenic key factor expression in mice. PLoS One. 2012;7(4):e35316.
16. Taura S, Taura M, Kamio A, Kummerow FA. Vitamin D-induced coronary atherosclerosis in normolipemic swine: comparison with human disease. Tohoku J Exp Med. 1979;129(1):9-16.
17. Semba RD. Vitamin A as “anti-infective therapy, 1920-1940. J Nutr. 1999;129(4):783-91.
18. Spiesman IG. Massive doses of vitamins A and D in the prevention of the common cold. Arch Otolaryngol. 1941;34(4):787-91.
19. Chitnis PR. Photosystem I: Function and Physiology. Annu Rev Plant Phsyiol Plant Mol Biol. 2001;51:593-626.
20. Han S, Chen LS, Jiang HX, Smith BR, Yang LT, Xie CY. Boron deficiency decreases growth and photosynthesis, and increases starch and hexoses in leaves of citrus seedlings. J Plant Physiol. 2008;165(13):1331-41.
21. Henry KM and Kon SK. The vitamin D content of English butter fat throughout the year. Biochem J. 1942;36(5-6):456-9.
22. Davies MJ, Fulton WF, Robertson WB. The relation of coronary thrombosis to ischaemic myocardial necrosis. J Pathol. 1979;127(2):99-110.
23. DeWood MA, Spres J, Notske R, Mouser LT, Burroughs R, Golden MS, Lang HT. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N Engl J Med. 1980;303(16):897-902.
24. Libby P and Theroux P. Pathophysiology of Coronary Artery Disease. Circulation. 2005;111:3481-8.
25. Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371-5.
26. Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103(7):934-40.
27. Wang L, Song Y, Manson JE, Pilz S, Marz W, Michaelsson K, Lundgvist A, Jassal SK, Barett-Connor E, Zhang C, Eaton CB, May HT, Anderson JL, Sesso HD. Circulating 25-hydroxy-vitamin D and risk of cardiovascular disease: a meta-analysis of prospective studies. Circ Cardiovasc Qual Outcomes. 2012;5(6):819-29.
28. Zittermann A, Kuhn J, Dreier J, Knabble C, Gummert JF, Borgermann J. Vitamin D status and the risk of major adverse cardiac and cerebrovascular events in cardiac surgery. Eur Heart J. 2013;34(18):1358-64.
29. Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69(5):842-56.
30. Toups MA, Kitchen A, Light JE, Reed DL. Origin of Clothing Lice Indicates Early Clothing Use by Anatomically Modern Humans in Africa. Mol Biol. Evol. 2011;28(1):29-32.
31. Elias PM and Williams ML. Re-apparaisal of current theories for the development and loss of epidermal pigmentation in hominids and modern humans. J Hum Evol. 2013;64:687-92.
32. Barham LS. Systematic Pigment Use in the Middle Pleistocene of South-Central Africa. Current Anthropology. 2002;43(1):181-90.
33. Wheeler PE. The thermoregulatory advantages of heat storage and shade-seeking behavior to hominids foraging in equatorial savannah environments. Journal of Human Evolution. 1994;26:339-350.
34. Luxwolda MF, Kuipers RS, Kema IP, Dijck-Brouwer DAJ, Muskiet FAJ. Traditionally living populations in East Africa have a mean serum 25-hydroxyvitamin D concentration of 115 nmol/L. BJN. 2012;108(9):1557-61.
35. Petit JR, Jouzel J, Raynaud D, Barkov NI, Barnola JM, Basile M, Bender J, Chappellaz M, Davis G, Delaygue G, Delmotte M, Kotlyakov VM, Legrand M, Lipenkov VY, Lorius C, Ritz C, Saltzman E, Stievenard M. Climate and atmospheric history of the past 420,000 years from the Vostok ice core, Antarctica. Nature. 1999;399:429-36.
36. Rejnmark L, Jorgensen ME, Pedersen MB, Hansen JC, Heickendorff L, Lauridsen AL, Mulvad G, Siggard C, Skjoldborg H, Sorensen TB, Pedersen EB, Mosekilde L. Vitamin D insufficiency in Greenlanders on a westernized fare: ethnic differences in calcitropic hormones between Greenlanders and Danes. Calcif Tissue Int. 2004;74(3):255-63.
37. Weaver CM, McCabe LD, McCabe GP, Braun M, Martin BR, Dimeglio LA, Peacock M. Vitamin D status and calcium metabolism in adolescent black and white girls on a range of controlled calcium intakes. J Clin Endocrinol Metab. 2008;93(10):3907-14.
38. Signorello LB, Shi J, Cai Q, Zheng W, Williams SM, Long J, Cohen SS, Li G, Hollish BW, Smith JR, Blot WJ. Common variation in vitamin D pathway genes predicts circulating 25-hydroxyvitamin D Levels among African Americans. PLoS One. 2011;6(12):e28623.
39. D’Amour P, Rousseau L, Hrnyak S, Yang Z, Cantor T. Influence of Secondary Hyperparathyroidism Induced by Low Dietary Calcium, Vitamin D Deficiency, and Renal Failure on Circulating Rat PTH Molecular Forms. Int J Endocrinol. 2011;469783.
40. Ternes SB, Rowling MJ. Vitamin D transport proteins megalin and disabled-2 are expressed in prostate and colon epithelial cells and are induced and activated by all-trans-retinoic acid. Nutr Cancer. 2013;65(6):900-7.
41. Waldron JL, Ashby HL, Cornes MP, Bechervaise J, Razavi C, Thomas OL, Chugh S, Deshpande S, Ford C, Gama R. Vitamin D: a negative acute phase reactant. J Clin Pathol. 2013;66(7):620-2.
42. Urbschat A, Paulus P, von Quernheim QF, Bruck P, Badenhoop K, Zeuzem S, Ramos-Lopez E. Vitamin D hydroxylases CYP2R1, CYP27B1 and CYP24A1 in renal cell carcinoma. Eur J Clin Invest. 2013;43(12):1282-90.
This article appeared in Wise Traditions in Food, Farming and the Healing Arts, the quarterly journal of the Weston A. Price Foundation, Winter 2013.
🖨️ Print post
The number of data points above 40 in fig 1 make it impossible to make a call on Vit D levels higher than 40 and there is no reference to sample sizes within fig 2 and the high levels of vit D.